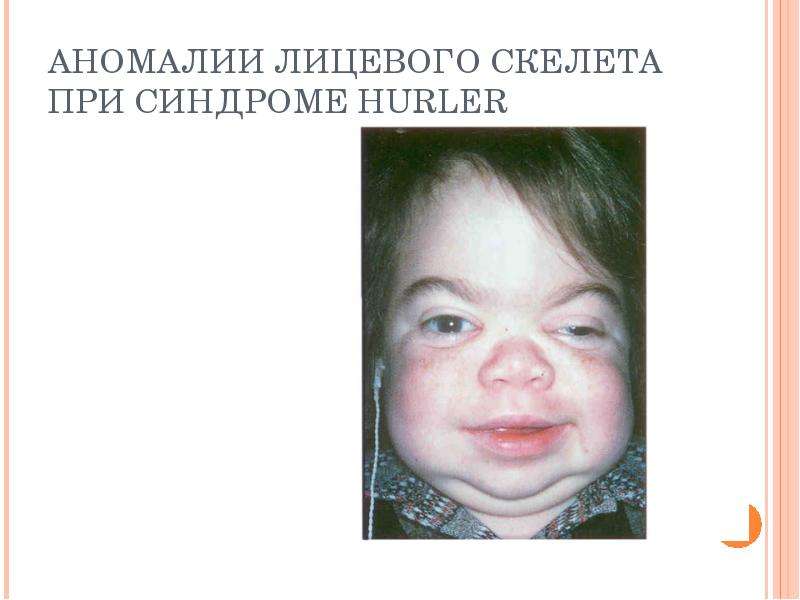
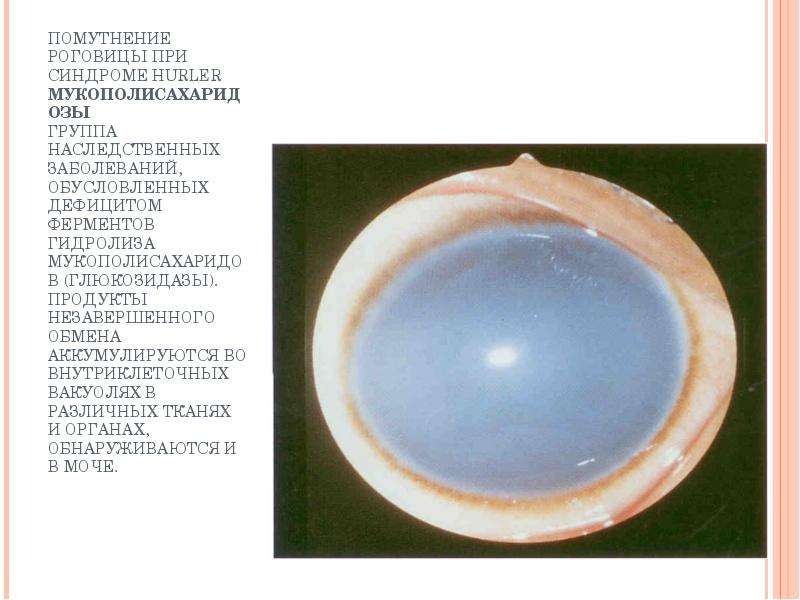
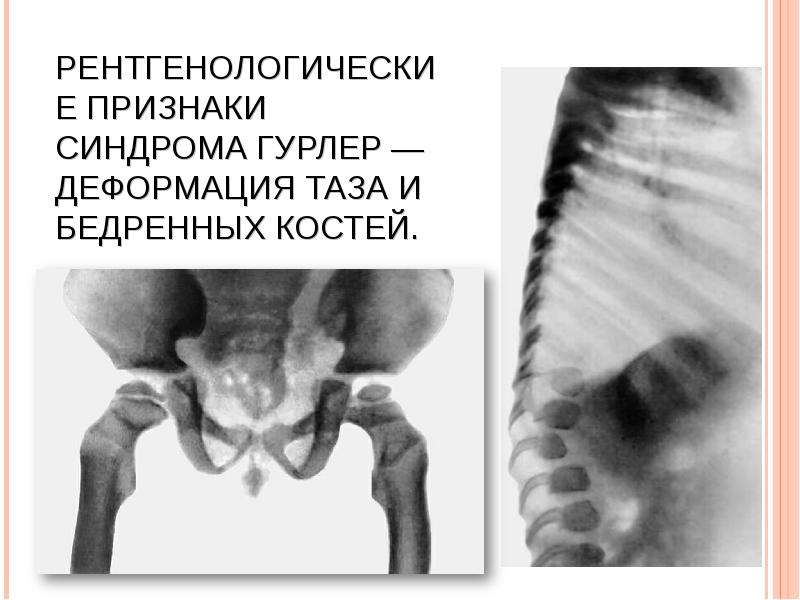
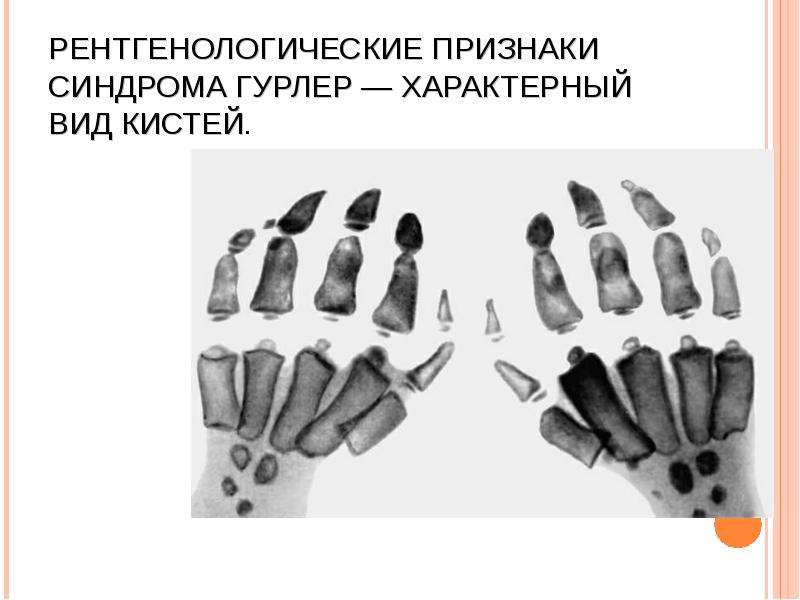
Описание слайда:
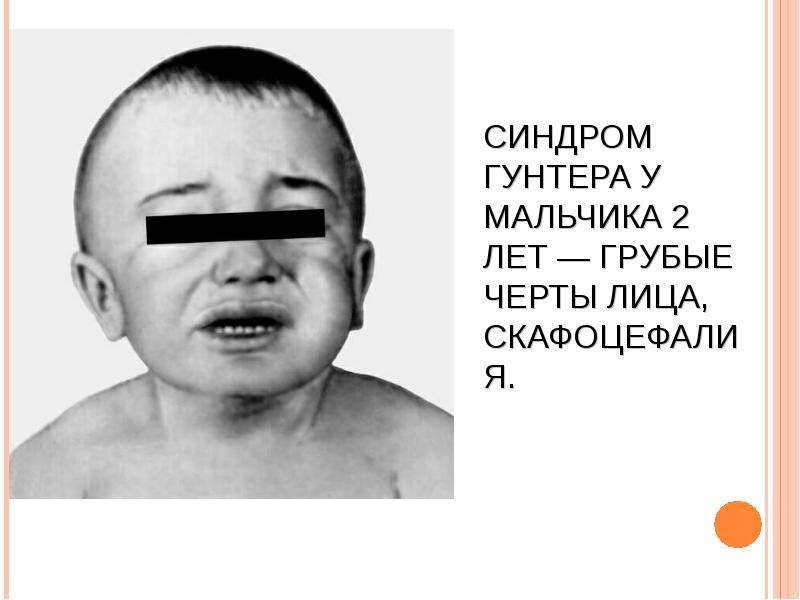
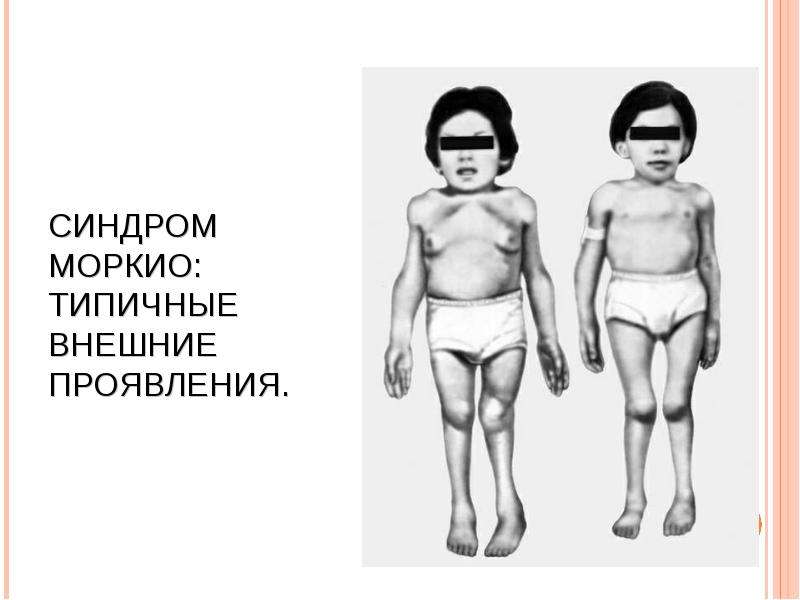
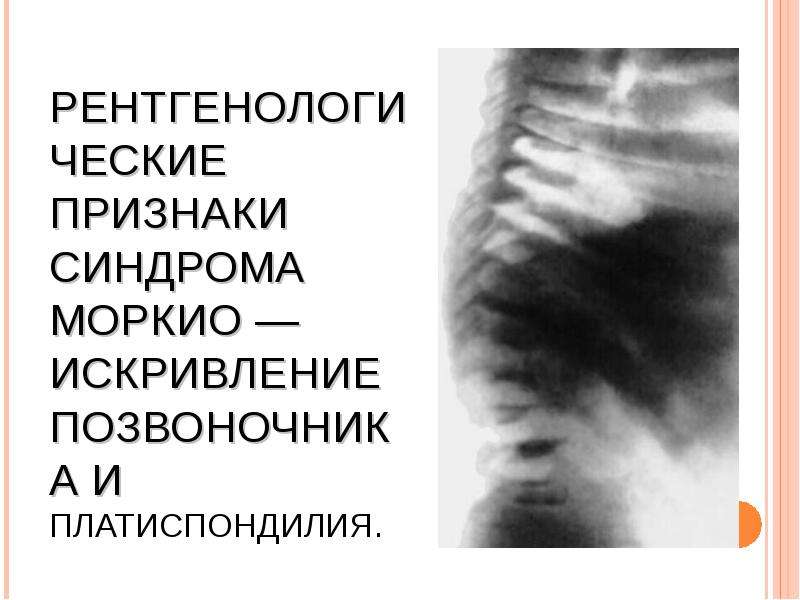
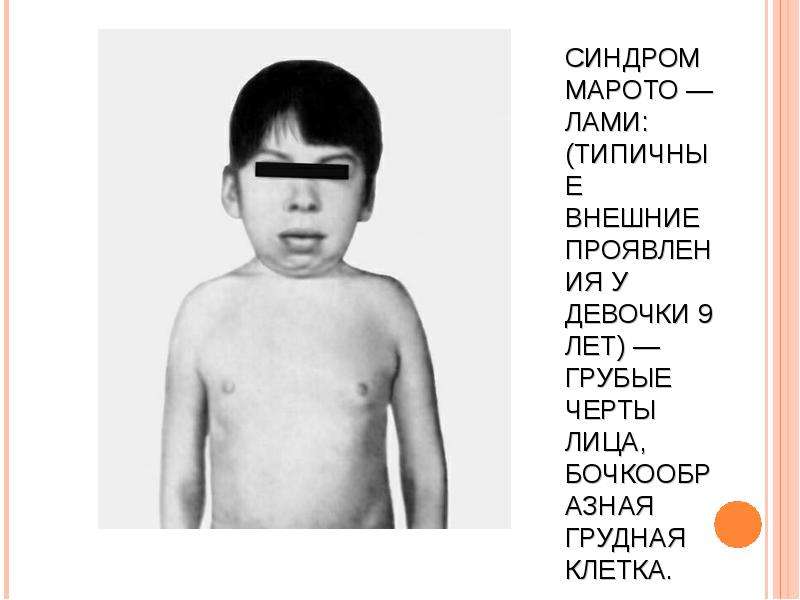
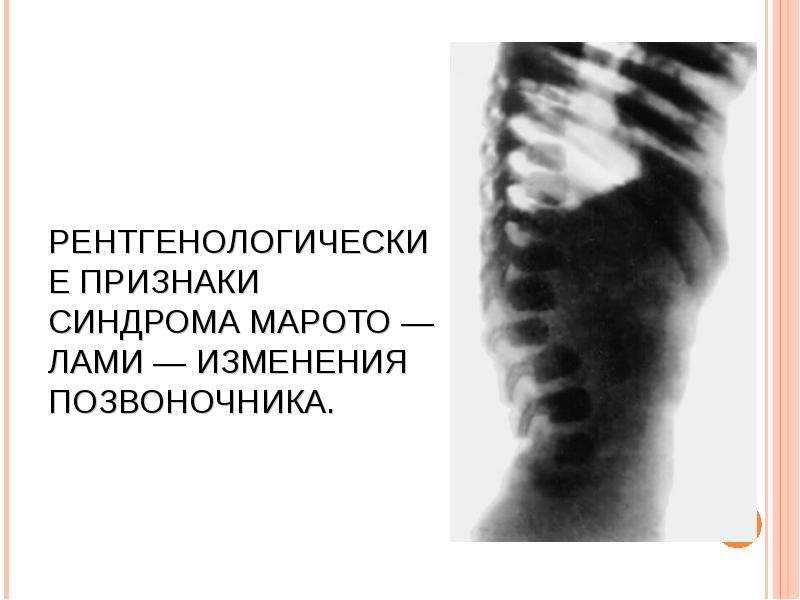
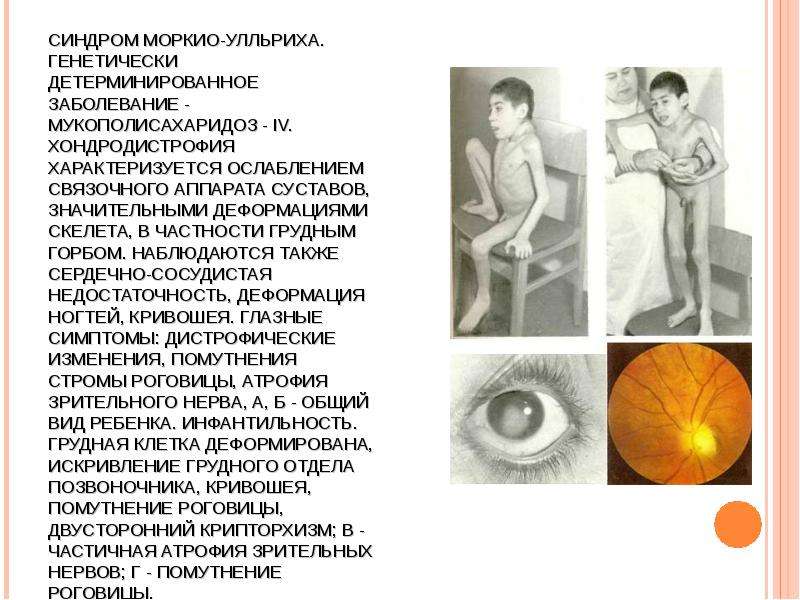
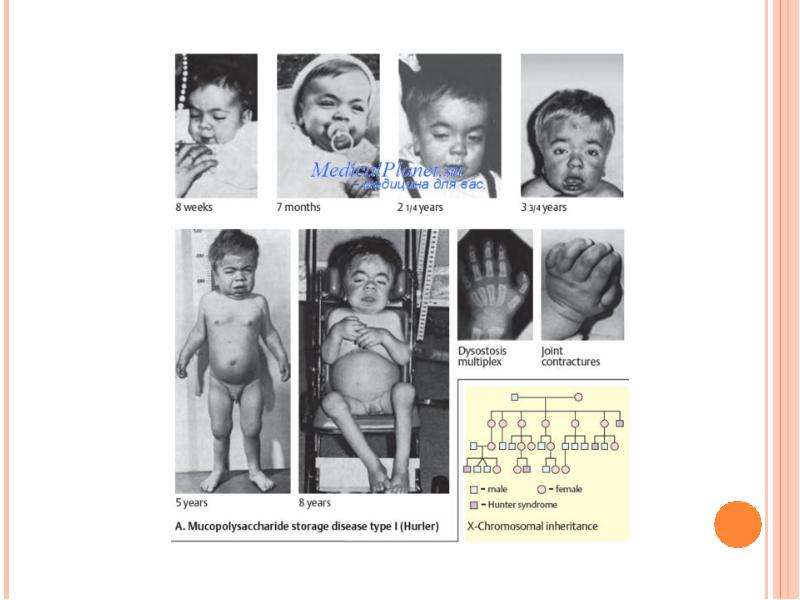
МУКОПОЛИСАХАРИДОЗ ТИПА IV (СНИДРОМ МОРКИО, БОЛЕЗНЬ МОРКИО). Заболевание в 1929 г. независимо друг от друга впервые описали уругвайский педиатр Моркио (L. Morquio) и английский радиолог Брейлсфорд (J.F. Braiisford). Частота до 1: 40 000. Дети рождаются без признаков болезни. Первые симптомы появляются в возрасте 1—3 года, и к 7—8 годам клиническая картина уже полностью выражена. Отмечаются резкая задержка роста (рост взрослого больного около 100 см), непропорциональное телосложение (относительно короткое туловище, микроцефалия, короткая шея), грубые черты лица, деформация грудной клетки (куриная, бочкообразная, килеобразная), кифоз или сколиоз грудного и поясничного отделов позвоночника (рис. 5). Питание снижено. Возникают контрактуры в локтевых, плечевых, коленных суставах, отмечается вальгусная деформация нижних конечностей, плоскостопие. Мышечная сила снижена. В случае компрессии спинного мозга помимо мышечной гипотонии отмечается поражение пирамидной системы, возможно развитие параплегии, паралича дыхания. Кожа утолщена, ее тургор и эластичность снижены. Часто выявляются пупочные и паховые грыжи, расхождение прямых мышц живота. Нередко отмечается снижение слуха, дистрофические процессы в роговице. Почти у всех больных, доживших до 20 лет, развивается глухота. Интеллект не снижен. При рентгенологическом исследовании обнаруживаются изменения позвонков: в шейном отделе выявляется гипоплазия или аплазия зубовидного отростка, в грудном — сколиоз, поясничном — кифоз; во всех отделах отмечается платиспондилия — уплощение и расширение тел позвонков, чем объясняется характерное укорочение туловища и необычно короткая шея. Изменяются кости таза: вертлужные впадины плоские и широкие, их крыша скошена, крылья подвздошных костей неправильной формы; контуры всех костей неровные; головки бедренных костей уплощены. Характерна увеличивающаяся с возрастом вальгусная деформация нижних конечностей. Пястные кости и фаланги укорочены и утолщены; проксимальные концы пястных костей конусообразные, концевые фаланги гипоплазированы . Кости предплечья укорочены; локтевая кость не достигает луче-запястного сустава, отмечается вывих ее головки в локтевом суставе; эпифизы треугольной формы. Дистальные эпифизы костей голени скошены, стопы деформированы. Дифференциальный диагноз проводят с различными вариантами нанизма, при котором отсутствуют специфические изменения скелета. В большинстве случаев летальный исход наступает до 20 лет вследствие сердечно-легочной недостаточности, развивающейся на фоне интеркуррентных заболеваний. Возможна внезапная смерть в результате смещения атланто-окципитального сочленения и повреждения ствола мозга.